Introduction
A CDSCO or USFDA inspection is not the moment to discover a gap it is the moment a gap becomes a warning letter, an import alert, or a licence suspension. Once an inspector is on the floor, findings stop being theoretical. They become Form 483 observations, NSQ classifications, or show-cause notices with fixed deadlines. Most of these findings are avoidable they surface because self-inspection was weak or documentation was reviewed only on paper, never tested on the floor. A mock audit closes this gap. It replicates a real inspection walkthrough, documentation pull, data integrity checks, interviews before the real one happens, while correction is still possible.
IMARC Engineering conducts pharmaceutical mock audits, GMP gap assessments, and regulatory compliance verification for manufacturers preparing for CDSCO, WHO-GMP, and USFDA inspections across India. This article draws on that work to explain what a mock audit should cover, why regulatory scrutiny is intensifying, and how a structured self-inspection programme prevents avoidable enforcement action.
Why Mock Audits Have Become a Strategic Compliance Tool
Pharmaceutical manufacturers no longer conduct mock audits solely to prepare for regulatory inspections. Today, mock audits are integrated into broader quality management systems to strengthen data integrity, improve operational discipline, identify systemic compliance gaps, and support continuous improvement. As regulatory expectations continue to evolve under revised Schedule M and international GMP standards, routine mock audits help organizations maintain inspection readiness throughout the year rather than only before scheduled inspections.
Why Regulatory Scrutiny Is Intensifying for Indian Pharmaceutical Manufacturers
Several data points, all from official regulatory sources, point to the same trend enforcement is tightening faster than most facilities are upgrading.
Rising Non-Compliance Findings
- CDSCO classified 1,879 drug batches as Not of Standard Quality in 2025, more than double the 877 batches recorded in 2024, per CDSCO data.
USFDA Enforcement Is Accelerating
- USFDA issued 73% more warning letters to pharmaceutical manufacturers in H2 2025 than in H2 2024, per industry tracking of USFDA disclosures.
- Import alerts and warning letters to Indian sites stood at 23 in the first eleven months of 2025, consistent with 25 in 2023 and 21 in 2024, per ICRA.
- USFDA conducted 212 inspections of Indian drug facilities in 2025, against 284 in 2024; No Action Indicated outcomes fell from 158 to 109.
The Schedule M Compliance Gap
- Of India’s approximately 10,500 pharmaceutical manufacturing units, only around 2,000 currently hold WHO-GMP certification, leaving more than 8,000 units at varying stages of the revised Schedule M gap, per industry compliance data.
- Large manufacturers (turnover above ₹250 crore) were required to comply with revised Schedule M by June 2024; MSMEs that formally applied for an extension have until December 31, 2025, per CDSCO notifications.
Export Stakes Keep Rising
- India’s pharmaceutical exports crossed US$31 billion in FY2025–26, up from US$15.44 billion a decade earlier, per Pharmexcil.
- India supplies 57% of WHO-prequalified APIs globally and hosts 670 USFDA-approved manufacturing facilities more than any country outside the United States.
A Form 483 Rarely Stays a Form 483
- A Form 483 carries a more than 50% chance of escalating into a warning letter when a company’s response is inadequate, per a peer-reviewed review of USFDA inspection data.
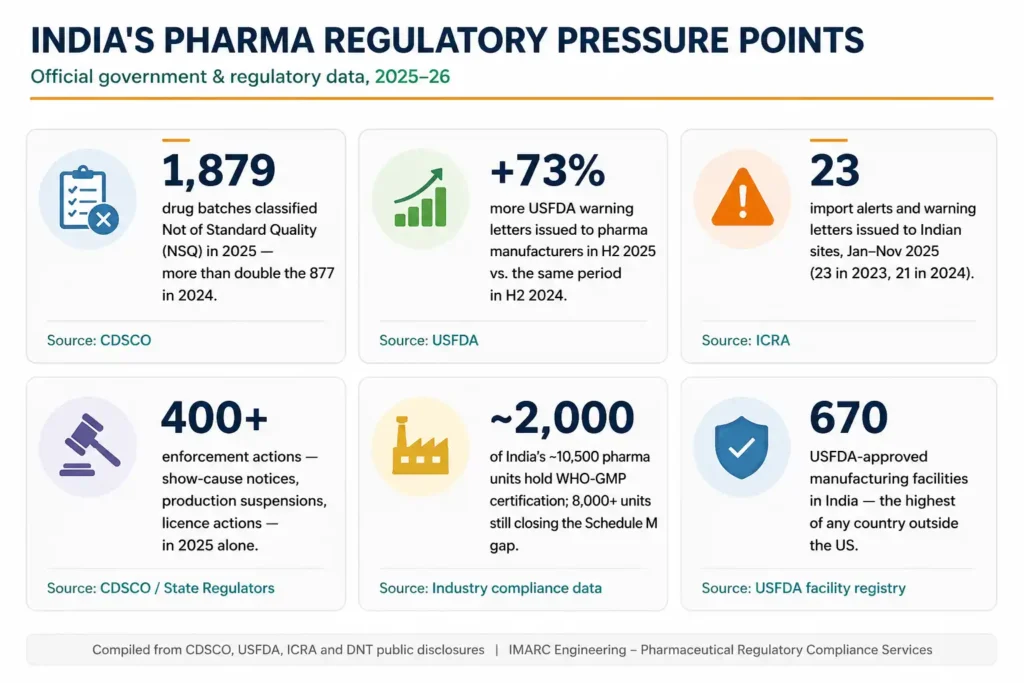
Figure 1: India’s Pharma Regulatory Pressure Points, 2025–26 CDSCO, USFDA, ICRA data
Why Skipping Mock Audits Creates Risk That Is Difficult to Unwind
The categories of risk that surface after an unprepared inspection:
- Import alert exposure: An OAI classification can block most shipments to the US, as a major Gujarat facility’s 2025 import alert showed.
- NSQ escalation: Batches classified Not of Standard Quality trigger recall obligations and customer disqualification, not just a compliance note.
- Licence action: Non-compliant manufacturers face show-cause notices, production suspension, and facility closure under active CDSCO enforcement.
- Requalification delay: An unprepared facility often needs 6–12 months of remediation before regulators reconsider approval.
- Remediation cost: Corrective action after a serious finding commonly runs ₹2–10 crore per facility, excluding lost production.
- Reputational loss: A public warning letter is visible to every competitor and customer evaluating the same facility.
Structured mock audits surface these risks or their absence while the manufacturer still controls the outcome.
Key Areas a Pharmaceutical Mock Audit Should Cover
A credible mock audit examines six interconnected areas drawn directly from Schedule M, WHO-GMP, and USFDA cGMP expectations.
1. Documentation & Data Integrity
- ALCOA+ compliance across batch records, eBMR, and LIMS
- Audit trail review for all GMP-relevant computer systems
- Raw data reconciliation against final reports
2. Quality Systems & CAPA
- CAPA closure timelines and root-cause depth, not just corrective action
- OOS and OOT investigation quality
- Quality Unit’s authority to stop production, not just document it
3. Manufacturing & Facility Controls
- HVAC, water system, and utility qualification status
- Equipment calibration and cleaning validation records
- Housekeeping and material flow discipline on the floor
4. Regulatory & Licence Compliance
- Factory licence and revised Schedule M compliance status
- Environmental consents (CTE/CTO) under CPCB classification
- WHO-GMP certificate and product-specific approvals (CDSCO, BIS, FSSAI as applicable)
5. Personnel & Training Readiness
- Technical staff qualifications and SOP training records
- Interview readiness can operators explain the process, not just perform it
6. Self-Inspection Programme Maturity
- WHO guidance calls for full self-inspection coverage at least once a year; Schedule M, Part I, Section 15 requires a documented internal audit programme
- Independence of the audit team from the area being audited
- Whether findings actually change practice, or repeat year after year

How IMARC Engineering Conducts Pharmaceutical Mock Audits
IMARC Engineering’s mock audit process is built to mirror an actual CDSCO or USFDA inspection, not a documentation checklist.
- Facility & documentation walkthrough: On-site review of premises, utilities, and batch records as a regulatory inspector would encounter them.
- Data integrity & CSV review: ALCOA+ testing of electronic records, audit trails, and computer system validation status.
- Quality systems assessment: Evaluation of CAPA history, OOS investigations, and Quality Unit authority on the floor.
- Regulatory compliance cross-check: Verification of licences, consents, and product approvals against source documents, not self-declaration.
- Gap report & CAPA roadmap: Findings graded critical, major, or minor, with named owners and target closure dates.
- Follow-up verification audit: A second-pass review to confirm CAPA effectiveness before the real inspection.
IMARC Engineering supports mock audits across pharmaceuticals, food and beverage, chemicals, and medical devices, where inspection expectations differ by category.
Speak with IMARC Engineering’s Pharmaceutical Mock Audit and GMP Compliance Specialists:https://www.imarcengineering.com/contact?service=sop-compliance-and-mock-inspections

Common Mistakes in Mock Audit Preparation
- Treating it as a paperwork exercise: Reviewing documents in a conference room without a floor walkthrough misses what inspectors look for first.
- Auditing your own area: Self-inspection without independent auditors produces findings that mirror what the department already believes.
- Ignoring data integrity: CSV and audit trail review are now central to both revised Schedule M and USFDA cGMP skipping them leaves the top enforcement trigger unchecked.
- No CAPA tracking discipline: Findings with no owner, deadline, or verification step repeat the same gaps the following year.
- One-time audit instead of a programme: WHO and Schedule M both expect periodic, risk-based coverage, not a single pre-inspection scramble.
- Under-scoping the audit: Skipping warehousing, QC labs, and pharmacovigilance leaves blind spots inspectors will find anyway.
Conclusion
As regulatory expectations continue to evolve, pharmaceutical manufacturers need a structured approach to identify and resolve compliance gaps before official inspections. Pharmaceutical mock audits strengthen inspection readiness, improve documentation quality, and reduce the risk of costly regulatory actions while supporting long-term quality and compliance.
As pharmaceutical manufacturing becomes increasingly regulated and export-oriented, inspection readiness is evolving into a continuous operational requirement rather than a periodic compliance exercise. Organizations that integrate structured mock audits, data integrity reviews, and proactive CAPA management into their quality systems are better positioned to meet regulatory expectations while strengthening long-term manufacturing performance.
Frequently Asked Questions
What is a pharmaceutical mock audit?
A planned internal inspection that replicates a CDSCO, WHO-GMP, or USFDA audit documentation, walkthrough, data integrity, and interviews before the real inspection happens.
How often should mock audits be conducted?
WHO guidance recommends full self-inspection coverage at least once a year, with higher-risk areas reviewed more frequently on a risk-based schedule.
What is the difference between a mock audit and a routine CDSCO inspection?
A mock audit is voluntary and internally controlled, giving time to fix findings. A CDSCO or USFDA inspection is regulatory and carries immediate consequences, including Form 483s or licence action.
Can a mock audit prevent a USFDA warning letter?
It cannot guarantee an outcome, but it substantially reduces the risk by finding the same gaps most warning letters cite before a regulator does.
Is a mock audit mandatory under Indian law?
Self-inspection is mandated under Schedule M, Part I, Section 15; the specific format and frequency of a mock audit follows the manufacturer’s own risk-based programme.
Contact Us:
IMARC Engineering
Phone: +91-120-433-0800
Email: sales@imarcengineering.com
India: C-130, Sector 2, Noida, Uttar Pradesh 201301
LinkedIn: https://www.linkedin.com/showcase/imarc-engineering/