
Introduction
In India, a CDMO or CMO relationship is a regulated manufacturing partnership not a vendor arrangement. Under Schedule M of the Drugs and Cosmetics Rules, regulated by the Central Drugs Standard Control Organisation (CDSCO), contract manufacturers are responsible for maintaining GMP compliance, quality systems, and product integrity throughout the manufacturing lifecycle.
For pharmaceutical companies looking to outsource manufacturing, choosing a reliable Pharmaceutical CDMO/CMO Partner in India is one of the most important strategic decisions. The right partner can accelerate product development, ensure regulatory compliance, and support long-term growth, while the wrong choice can create costly operational and quality challenges.
CDMO/CMO selection failures follow predictable patterns: price-led decisions, certificate acceptance without inspection history review, no technology transfer assessment, and no supply chain risk evaluation.
The seven checks in this advisory are the structured evaluation framework quality assurance professionals, procurement teams, and investment evaluators must apply before any CDMO/CMO selection is finalised and before a single clause of a manufacturing services agreement is signed.
Why CDMO/CMO Selection Failures Are Increasing
According to the Department of Pharmaceuticals, Government of India, India’s pharmaceutical industry contributes approximately ₹4.5 lakh crore in annual production value, per Department of Pharmaceuticals data with exports exceeding ₹2.06 lakh crore annually. This growth is feeding more CDMO/CMO relationships but not proportionally more evaluation rigour.
- Data published by the Central Drugs Standard Control Organisation (CDSCO) indicates, 500+ new pharmaceutical manufacturing site approvals annually under CDSCO expanding the CDMO/CMO market faster than quality differentiation tools can keep pace
- US FDA issues Form 483 observations in 50–60% of inspections of Indian pharmaceutical facilities significantly higher than the global average, per FDA’s inspection database
- Technology transfer failures add an average of 6–18 months to commercial launch timelines when they occur
- A single product recall traced to a CMO quality failure can cost a sponsor ₹50 crore or more in product destruction, market withdrawal, and regulatory remediation
- An active US FDA Import Alert takes 18–36 months to resolve during which US market supply is interrupted
The Most Common CDMO/CMO Selection Mistakes
- Selecting on price alone: a CDMO offering 20–25% lower processing fees may carry US FDA 483 observations, QMS gaps, or technology transfer limitations whose downstream cost far exceeds the initial saving
- Certificate acceptance without inspection history review : a WHO-GMP or US FDA certificate does not disclose the observation history, outstanding CAPA commitments, or last inspection date
- No technology transfer capability assessment : sponsors sign manufacturing agreements based on dosage form capability claims, without evaluating documented TT protocols or scale-up track records
- Ignoring scalability : a CDMO suited for clinical supply may become a structural bottleneck at peak commercial demand
- Inadequate supply chain risk assessment : single-source API dependencies and no business continuity plan are risks that transfer to the sponsor the moment a manufacturing agreement is signed.

Check 1: Regulatory Compliance and Certification Status
Regulatory compliance is the first and non-negotiable screening criterion. It establishes the legal baseline for which markets a CDMO can supply and which it cannot.
What to Verify
- WHO-GMP : baseline for regulated emerging markets; verify currency on WHO Prequalification database, not from CDMO-supplied copies
- US FDA : mandatory for US market supply under 21 CFR Parts 207, 210, and 211; facility inspection history searchable on FDA’s Establishment Inspection Report database
- EU GMP : required for European market supply; verify facility status on EMA’s EudraGMP database
- MHRA : governs UK supply post-Brexit; separate from EU GMP; requires valid MHRA GMP certificate
- TGA : Australian market requires TGA GMP clearance; not interchangeable with EU GMP or WHO-GMP
Beyond the Certificate
- Inspection history date of last inspection, issuing authority, and number of observations raised
- Warning letters and Import Alerts searchable on FDA’s website; any active warning letter is a disqualification criterion
- Outstanding CAPA commitments to regulatory authorities none should be open
- Re-inspection timeline a facility not inspected for 3+ years carries unknown current compliance status
Per US FDA’s inspection database, Indian pharmaceutical manufacturing facilities received over 100 warning letters in a five-year period. Sponsors who sign agreements with warning-letter facilities inherit the supply risk those letters represent.
Check 2: Manufacturing Capabilities and Technical Expertise
A certificate verifies compliance at inspection. It does not verify that the facility can manufacture your specific product.
- Dosage form capability: oral solids, sterile injectables, aseptic fill-finish, lyophilised products, modified-release formulations, and biologics each require fundamentally different facility design and operational expertise
- API contract manufacturing : ICH Q7 compliance governs API GMP; containment capability for high-potency compounds (OEB classification) must be verified
- Equipment qualification status: IQ/OQ/PQ records for all critical equipment, utility system validation (purified water, HVAC, clean room classification), and preventive maintenance logs are mandatory review items
- Specialised technology claims : modified-release platforms, continuous manufacturing, and controlled substance handling under NDPS Act compliance each require production history evidence not capability claims
Evaluation standard: Request batch manufacturing records for at least 3 commercial batches of a product comparable in complexity to the one being outsourced.
Check 3: Quality Management Systems and Batch Consistency
The quality management system is the operational infrastructure through which compliance is sustained between regulatory inspections.
QMS Elements to Evaluate
- SOP governance : currency, version control, and staff training records; out-of-date or unsigned SOPs are a leading FDA 483 observation category
- CAPA system : the ratio of open to closed CAPAs over 12 months is a quantitative indicator of QMS discipline
- Deviation management : volume, classification (minor/major/critical), and closure rate; high deviation volume with rapid closure is better than low reported volume, which typically indicates under-reporting
- Change control : weak change control is consistently cited in FDA warning letters as a root cause of product quality failures
Batch-to-Batch Consistency
- Request batch release data across 12–18 months of production history
- OOS rates below 0.5% are expected for well-controlled solid oral dose manufacturing; rates above 1% warrant investigation
Per CDSCO inspection data, quality management system deficiencies including inadequate CAPA systems and SOP non-compliance are among the most frequently cited GMP violations in Indian pharmaceutical manufacturing facilities during joint inspections with international regulatory authorities.
Check 4: Production Capacity and Scalability
Capacity evaluation must account for where the product needs to be in 3–5 years not only where it is today.
- Current utilisation : a facility operating at 85–90% installed capacity cannot reliably absorb a new programme; request a current utilisation report by product type and manufacturing line
- Expansion capability : site master plans, historical capex records, and expansion approval status under CDSCO and state drug licensing authorities indicate whether a CDMO can grow with your product
- Supply continuity risk : what happens if a key manufacturing line is taken out of service? What if the CDMO’s key technical personnel depart? Scenario assessment is required, not only baseline capacity verification
- Customer concentration : a CDMO over-dependent on a single major customer creates cross-subsidy risk that transfers to all other clients on their batch schedule
Check 5: Technology Transfer and R&D Support
Technology transfer capability is the most underweighted evaluation criterion in CDMO selection and the most frequently cited cause of post-contract relationship failures.
What a Successful Technology Transfer Requires
- Process development expertise : understanding of critical quality attributes (CQAs) and critical process parameters (CPPs) per ICH Q8
- Analytical method transfer : establishing analytical equivalence between sponsor reference methods and CDMO in-house testing protocols per ICH Q2
- Scale-up track record : documented history of successful transfers from lab scale to pilot batch to commercial production, with timeline data and issue resolution records
What Distinguishes Capable CDMOs
- Dedicated TT teams with formal protocols aligned to ICH Q10
- Documented transfer timelines with objective success metrics
- Non-capable CDMOs manage TT through the same personnel responsible for routine manufacturing with no formal methodology and no independent verification of transfer success
Evaluation standard: Request a documented technology transfer case study including timeline, issues during scale-up, and resolution methodology for at least two products comparable in complexity to the one being outsourced.
Check 6: Supply Chain Reliability and Risk Management
A pharmaceutical contract manufacturer’s operational reliability is a direct function of the supply chain it manages.
- Raw material sourcing risk : a significant proportion of starting materials for Indian formulation manufacturers are sourced from a limited number of manufacturing clusters, creating structural single-source dependencies; supply disruptions have resulted in interruptions of 3–9 months
- Supplier qualification : Approved Vendor List with active supplier audit programme; alternative sourcing established and validated for critical starting materials
- Business continuity planning : request the CDMO’s documented BCP; verify whether it has been formally tested; understand recovery time objectives for critical manufacturing lines
Per ICH Q10 and EMA’s Guidelines on Outsourced Activities, a contract manufacturer is expected to maintain a quality agreement addressing supply chain responsibilities including raw material sourcing risk, supplier qualification, and business continuity provisions that protect the sponsor’s supply continuity obligations.
Check 7: Commercial Terms, IP Protection, and Due Diligence Framework
Technical excellence without commercial clarity produces partnerships that are operationally sound but commercially unmanageable.
- Pricing transparency : itemised pricing structures with clear cost-pass-through mechanisms for raw material movements are a minimum requirement; all-in aggregated quotations create commercial opacity
- IP protection : confidentiality agreements, IP assignment clauses, non-compete provisions covering employees who access proprietary technology, and cybersecurity protocols require legal review before agreement execution
- Performance KPIs : batch release timelines, right-first-time manufacturing rates, OOS investigation response timelines, and audit readiness commitments must be contractually defined; a CDMO that resists defining KPIs is communicating how it intends to manage performance disputes
- Contract exit provisions : notice periods, technology transfer obligations on exit, analytical method ownership, and batch record retention are non-trivial to negotiate after a relationship has deteriorated
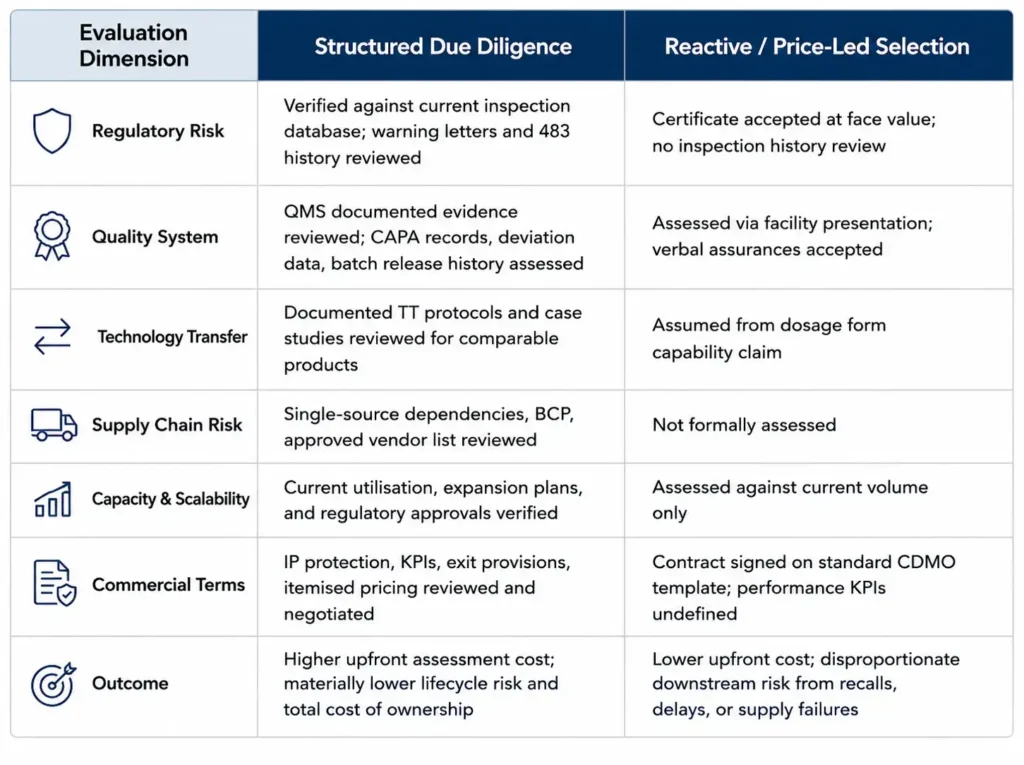
CDMO/CMO Evaluation: Structured Due Diligence vs Reactive Selection
How IMARC Engineering Supports Pharmaceutical CDMO/CMO Identification and Selection in India
IMARC Engineering provides end-to-end pharmaceutical CDMO/CMO identification, technical due diligence, and supplier qualification services for global pharmaceutical companies, biotech firms, and API manufacturers establishing or expanding manufacturing partnerships in India.
- Manufacturer identification structured identification of WHO-GMP, US FDA, EU GMP, and MHRA-approved facilities across oral solids, sterile injectables, API manufacturing, and specialised dosage forms, matched against client-specific product, regulatory market, and commercial requirements
- Technical due diligence independent assessment of facility capability, QMS maturity, technology transfer infrastructure, and supply chain risk; delivered as a risk-rated report with specific findings and recommendations
- Regulatory compliance evaluation : independent verification against primary regulatory databases; inspection history analysis; gap assessment against the specific regulatory market requirements of the client’s target supply chain
- Supplier qualification support : documentation package preparation and audit support aligned with 21 CFR Part 211 Subpart B, EU GMP Chapter 7 on outsourced activities, and ICH Q10 quality system requirements
Contact Our Team : https://www.imarcengineering.com/contact?service=contract-manufacturer-identification
CDMO/CMO Selection Is a Strategic Manufacturing Decision Not a Procurement Exercise
The numbers are clear:
- US FDA issues Form 483 observations in 50–60% of inspections of Indian pharmaceutical facilities compliance risk is not theoretical
- Technology transfer failures add 6–18 months to commercial launch timelines, with revenue loss not recoverable through contractual remedies
- Product recalls cost sponsors ₹50 crore or more per incident in product destruction, market withdrawal, and regulatory remediation
- Single-source API supply disruptions have demonstrated interruption periods of 3–9 months, with full commercial impact falling on the sponsor
- Active US FDA warning letters take 18–36 months to resolve during which a facility cannot supply to the US market
Manufacturers who commission on schedule and avoid recall costs are not the ones who got lucky with their CDMO selection. They are the ones who treated CDMO evaluation as a technical, regulatory, and commercial due diligence function executed with the same rigour applied to any other strategic manufacturing decision.
The seven checks in this advisory provide the evaluation architecture for exactly that. For manufacturers who cannot absorb the cost of a wrong selection, structured technical due diligence is not optional it is a project requirement.
For information on IMARC Engineering’s pharmaceutical CDMO/CMO identification, technical due diligence, and supplier qualification services, visit imarcengineering.com/services/regulatory-approval-and-licensing
Contact Us:
IMARC Engineering
Phone: +91-120-433-0800
Email: sales@imarcengineering.com
India: C-130, Sector 2, Noida, Uttar Pradesh 201301
LinkedIn: https://www.linkedin.com/showcase/imarc-engineering/


